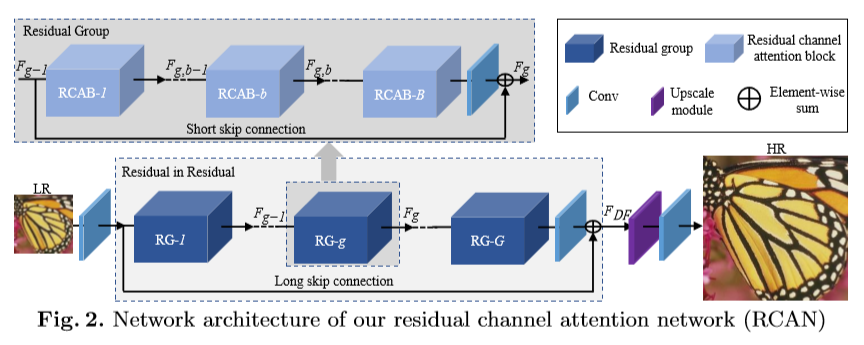
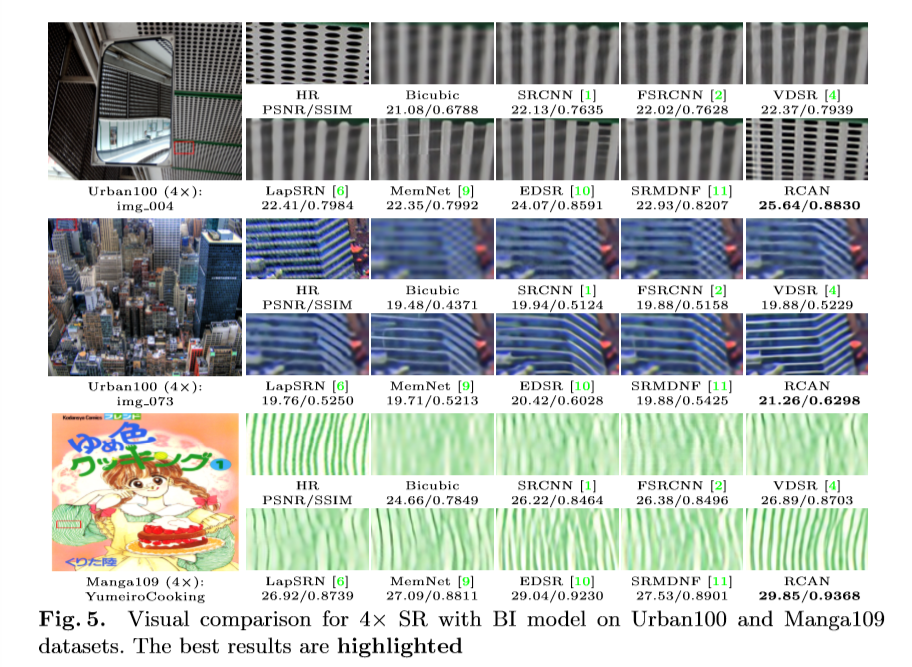
Currently in Ganapati’s lab, we are using Fourier Ptychographic Microscopy (FPM) to image biological samples. This type of microscopy involves using an LED dome to illuminate the sample in many different illumination angles, allowing for increased image information of the sample with both dark field and bright field images. However, we have come across some challenges with FPM when using thick or transparent biological samples.
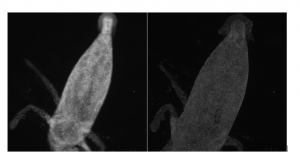
A different type of microscopy that is better suited for thick biological samples is fluorescence microscopy. In florescence microscopy, a sample is stained with various fluorophore dyes, with each dye attaching to different organic or mineral components of the sample. The stained sample is then bathed in intense light, causing the specimen to fluoresce. Because the specimen is now labeled with dyes, it is much easier to visually distinguish different layers of the sample in the image.
There are multiple types of super resolution algorithms that can be used for fourescence microscopy, but two in particular that this lab is interested in are structured light microscopy (SIM) and the point spread function (PSF).
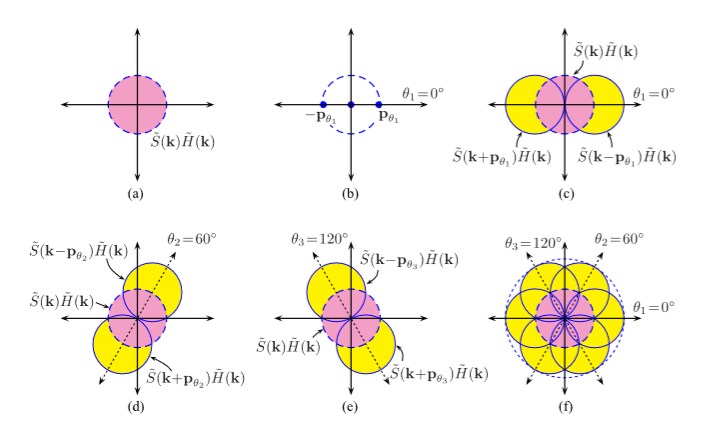
Structured Light Microscopy (SIM):
Point Spread Function (PSF):
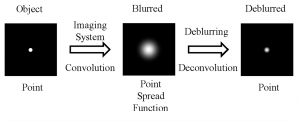
When you are viewing a microscopic fluorescent object under a microscope, the diffraction of light from the emitted fluorescence will cause the object to appear blurred. This blurring, which occurs in both the lateral and depth direction of the object, is known as the point spread function (PSF). PSF tend to have worse blurring in the depth direction, since microscopes tend to have worse resolution in that direction.
In order to minimize the effects of the PSF and reduce blurring, some modifications to the hardware of the microscope can be made, such as having a shorter wavelength of illumination light and increasing the numerical aperture of the objective lens. On the software side, computer algorithms can “undo” the blurring caused by the PSF by performing computational deconvolutions, which reverse the effects of the PSF and enhance the resolution of the image.
A Github repository for a PSF super resolution algorithm can be found here: https://github.com/FerreolS/tipi4icy
Resources:
A. Lal, C. Shan, and P. Xi . “Structured Ilumination Image Reconstruction Algorithm.” Department of Biomedical Engineering, Peking University. IEEE Journal. 4.(2016).
K. Sring and M. Davidson. “Introduction to Fluorescence Microscopy.” MicroscopyU.
“A Beginners Guide to the Point Spread Function”. Zeiss Microscopy.
A. Lavdas “Structured Illumination Microscopy–An Introduction.” Bitesizebio.com
N. Mansurov. “What is Moiré?” PhotographyLife.com (2012).